Meadowlark: Structure Processing Workflow
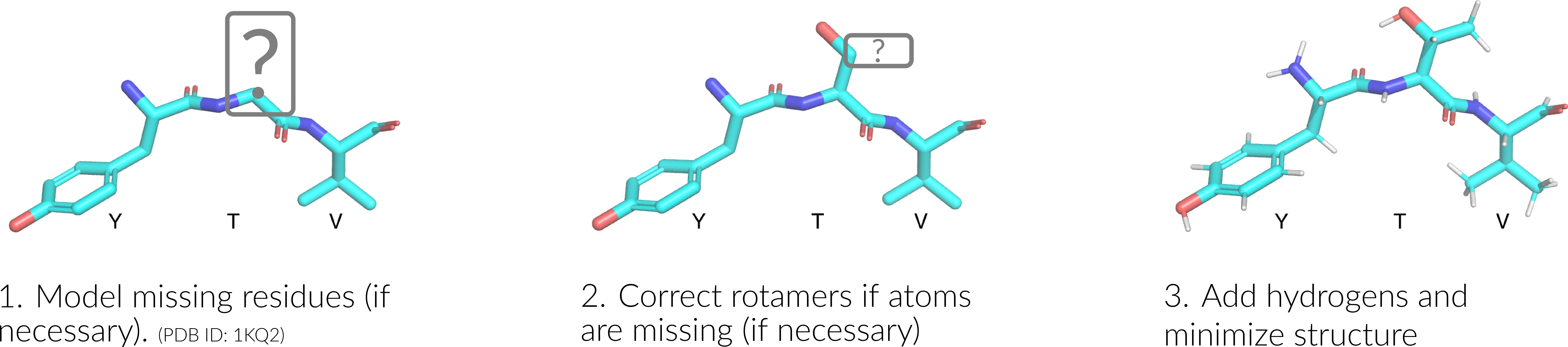
We ‘clean’ or ‘sanitize’ a starting protein structure by applying the following approach. We begin with coordinate-preserving tasks:
Select first model (from among multiple possible models in a PDB file)
Select the correct chain
Select the first alternate location (if multiple conformers are present for an atom/residue).
Remove hetero-atoms (water or buffer molecules, other crystallization reagents, etc.)
which are all achieved via pdb-tools (Q114840802).
Finally, we then performing coordinate-modifying tasks for each domain structure via the following stages:
Build/model any missing residues with MODELLER (Q3859815)
Correct/optimize rotamers with SCWRL4 (Q114840881), e.g. if there are any missing atoms
Add hydrogens and perform a rough potential energy minimization with the Pdb2Pqr (Q62856803).